
Understanding Risk Management Requirements in ISO 13485:2016
The current revision has new requirements for risk management. Here are some questions to consider and guidance for implementation.

ISO 13485 is the International Standard which outlines requirements for a Quality Management System (QMS) for Medical Devices. Developing and deploying a QMS that meets the requirements of this standard is necessary to commercialize your medical device in several important global markets.
ISO 13485 was revised in 2016 with a heavy emphasis on risk and planning. The term risk appears twice as many times in this revision compared to the prior 2003 version! Risk and Risk Management are now explicitly defined and a risk-based approach is required for control of appropriate processes in the QMS. A good understanding of these requirements is needed to ensure your QMS continues to be in compliance.
It is important to note that the requirement for a risk-based approach now appears very early in the standard, starting at the top-level general requirements for a QMS.
clause 4.1.2(b):
“The organization shall apply a risk-based approach to the control of appropriate processes needed for the quality management system.”
It all starts with the role your organization (clause 4.1.1) plays in the lifecycle of the medical device, and the processes you have identified and implemented in your QMS (4.1.2(a)). Control of these processes to achieve their desired results is now expected to be based on risk. In simple terms, high risk processes are expected to have a higher level of control compared to lower risk processes. This high-level requirement then flows down to specific requirements under different clauses related to personnel, suppliers, verification of externally provided products/services and validation/re-validation of software.
These newly added requirements can be difficult to interpret and implement. The following questions, and the accompanying discussion, are intended to help you evaluate the need for additional improvement actions in your QMS.
What is the scope of these risks? How to determine the level of risk?
It is important to understand that the term risk, within the context of ISO 13485, applies primarily to the safety and performance of the medical device. It also includes compliance to applicable regulatory requirements on a secondary basis. Business risks, while important for setting priorities and objectives, are not included in the scope of ISO 13485.
The term risk, as defined in ISO 13485, refers to the combination of the probability of occurrence of harm and the severity of that harm. This may cause some confusion in terms of the scope as the term harm is usually applicable to safety related issues. Keep in mind that the scope of ISO 13485 includes not only safety, but all product and applicable regulatory requirements. Therefore, risks related to product performance and regulatory compliance also need to be considered. For example, frequent service or maintenance issues may cause inconvenience to the user but not result in any harm to the patient beyond delay in treatment. As another example, CAPA effectiveness may be a regulatory compliance risk, but it may not directly result in any harm or damage. As a result, the risk-based approach required by the current revision includes safety, performance and compliance risks because all of them are required to be appropriately mitigated to ensure your QMS is effectively implemented and maintained.
Assessment of the risk level, whether high or low, depends on the combination of the severity of the undesired effects and the probability of their occurrence. You need to analyze each process of the QMS and identify various scenarios which may lead to these undesired effects on safety, performance and compliance. Next, you have to develop a system of rating each scenario for their probability and severity. While it is always preferred to have a numerical, quantitative scale for both probability and severity, it is not uncommon to use a qualitative scale if each level is clearly defined. In practice, everyone feels different about risk that is why it is important to establish a well-defined rating system and criteria for risk level.
You may already have a system for risk evaluation within your ISO 14971 risk management framework. It may just need to be modified to include risks related to performance and regulatory compliance.
What are the new requirements for risk management in the current revision of ISO 13485?
In addition to the stated requirement for risk management throughout product realization (Clause 7.1) in the 2003 version of the standard, the current revision has several newly added requirements in multiple clauses. A summary of these requirements with guidance for implementation is presented in the figure below.
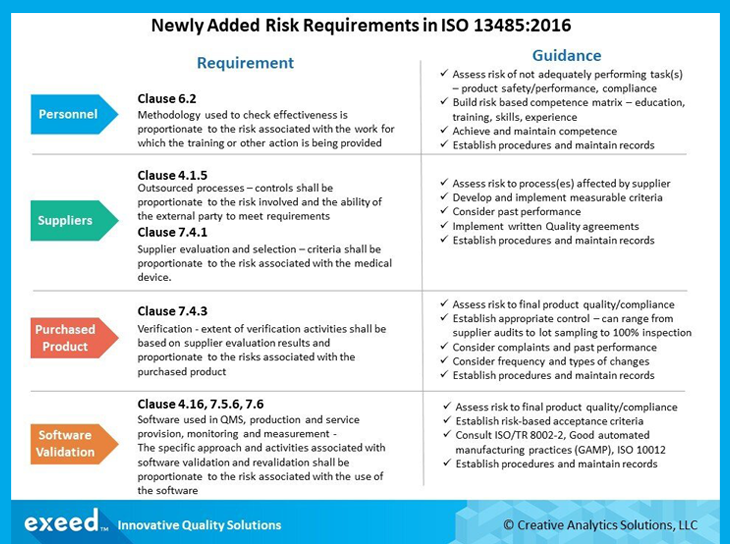
Clause 6.2 focuses on competence of personnel, which is based on the 4 pillars of education, training, skills and experience. In this context, the term competence implies demonstrated ability to complete a task and produce expected results. As a result, it is not just training, but skill and experience with the specific task that need to be demonstrated. A risk-based approach is now required to show that action(s) taken, including appropriate training and re-training, are effective in achieving and maintaining competence. The level of detail needed to establish effectiveness will be based on the impact of each task on risk to product safety and performance and compliance with applicable regulatory requirements.
Clauses 4.1.5 and 7.4.1 now require a risk-based approach for control of external providers for outsourced processes and other incoming products and/or services. Providers of products and services related to high risk processes will require a higher level of control in evaluation and selection. For example, criteria used to evaluate and select a third-party manufacturer of a critical part/raw material or sterilization process will need to be more rigorous than other non-critical items and/or processes. You may require them to have an ISO 13485 certified QMS, or ask for data to demonstrate high capability for critical safety and performance related parameters. Questions related to their reputation, business stability, credit rating etc. may also be relevant. The scope and level of required performance for evaluation and selection of external providers needs to be defined based on a risk assessment.
Clause 7.4.3 now requires that the extent of verification of purchased products (and services) be determined based on results of supplier evaluation and risk to final product quality and compliance. These activities may range from accepting on certificate of analysis (COA) to sampling to 100% inspection. Analysis of previous inspection data or customer complaints that may relate to purchased products may also be a factor in frequency and intensity of verification activities. History of responsiveness to nonconformances, supplier corrective action requests (SCAR) and effectiveness of implemented corrective/preventive actions may be mitigating factors. Frequency and types of changes in the supplier processes, their communication and evaluation should also be considered.
Clauses 4.1.6, 7.5.6 and 7.6 address risk requirements related to validation and re-validation of software used in the QMS processes, process validation and monitoring/measurement equipment. The extent of these activities needs to be proportionate to risk associated with the software in relation to product safety, performance and compliance with requirements. As an example, software used for process automated detection of nonconforming product will require a more extensive validation compared to software used for analyzing data related to QMS performance. It is useful to consult resources such as ISO/TR 80002-2 (validation of software for medical device quality management system), Good Automated Manufacturing Practices (GAMP) and ISO 10012 (Requirements for measurement processes and measuring equipment). As technology becomes an increasingly important aspect of medical device design and manufacturing, software validation needs to be an area of focus in your QMS. Another emerging area is software used for mitigation of cybersecurity risks, which also needs to be adequately validated to meet rapidly changing regulatory requirements.
Are there any other expectations for risk management which are not explicitly required in ISO 13485?
It is important to know what is explicitly required by the standard, and what will be expected in a more mature QMS. Here are a few examples of the application of the risk-based approach where ISO 13485 does not specifically outline risk consideration:
Clause 5.6 – Planned intervals for management reviews
Clause 7.5.1 – Control of production and service provision
Clause 8.3 – Handling nonconforming product and nature of required correction/corrective action
Clauses 8.5.2, 8.5.3 – Evaluating the need and nature of actions to prevent occurrence (preventive) and recurrence (corrective) of nonconformities
In general, wherever the term plan or planning is used in the standard, a risk-based approach is expected. Although there is no requirement for a formal risk management process, except in the specific clauses summarized in the figure above, it is reasonable to expect that this will gradually need to become a core competence of your Quality organization.
How to manage risk related requirements on an ongoing basis?
When thinking about implementing these risk related requirements, keep in mind that risks generally evolve as new information becomes available through post-market surveillance. Your QMS needs to be sufficiently resilient to respond to these rapidly evolving risks. There needs to be a mechanism to identify, assess and respond to new risks and/or to changes in the level of existing risks in terms of severity and probability of undesired effects. New controls may need to be implemented, or existing controls may need to be adjusted. If the QMS is designed using a process approach, with clearly defined processes and their interaction, it may be more resilient to changes in the risk profile of your products and regulatory requirements. The emphasis on planning throughout the current revision is intended to promote a more thoughtful, risk-based approach for maintaining the integrity and effectiveness of your QMS.
In conclusion, there is a heavy emphasis on a risk-based approach in the current revision of the ISO 13485 standard. There are several newly added requirements for risk management under different clauses related to personnel, suppliers, verification of externally provides products/services and validation/re-validation of software. It is important to fully understand these requirements so you can interpret them within the context of the role of your organization and your QMS processes.
Share your comments and questions below. Contact us and let us know how we can help.
References:
ISO 13485:2016 for Medical Devices, March 2016
ISO 13485:2016 – A Practical Guide, 2017
ISO 14971:2019 – Application of Risk Management to Medical Devices, December 2019
The Softer Side of Risk Management, September 2019
Cybersecurity is the Next Frontier of Risk Management, August 2019