
Understanding ISO 13485:2016
Exclusion vs. not applicable? How to make the right decision for your Quality Management System.

ISO 13485 is the International Standard which outlines requirements for a Quality Management System (QMS) for Medical Devices. Developing and deploying a QMS that meets the requirements of this standard is a critical step in getting market authorization for a medical device in many global markets.
ISO 13485 was revised in 2016 with several new requirements and a stronger emphasis on risk and planning. In fact, the older 2003 version did not define the term “risk” under Terms and Definition (Clause 3). A considerable amount of detail is now provided to clarify the concept of risk, which pertains to safety or performance requirements of the medical device or meeting applicable regulatory requirements within the scope of the revised standard.
While there are many differences between the 2003 version and the current 2016 revision, we will focus on 2 key topics in this blog. The first is the question of exclusion vs. non-applicability (Clause 1.0 – Scope), and the second is related to expectations on planning for product realization (Clause 7.1). Our interest in these topics was sparked by a robust discussion during a recent training session we provided to a diverse group of attendees in Atlanta. We feel it would be highly beneficial to clarify these topics and engage a broader audience in this discussion for further conversation.
Let us start with the topic of exclusion vs. non-applicable
Merriam-Webster dictionary defines the term exclusion (noun) as the act or an instance of excluding, or the state of being excluded. The verb exclude is defined as to prevent or restrict entrance of, or to bar from participation, consideration, or inclusion.
The term applicable is defined as capable of or suitable for being applied. The qualifier “not” with the term applicable would then change the definition to incapable of or unsuitable for being applied.
Let us now look at how ISO 13485:2016 uses this terminology in defining requirements.
Clause 1: Scope
“If applicable regulatory requirements permit exclusions of design and development controls, this can be used as a justification for their exclusion from the quality management system.”
Therefore, the entire Clause 7.3 – Design and Development, can be excluded from the QMS if allowed by applicable regulatory requirements. As an example, all but a few of Class I devices are exempt from Design Controls in 21 CFR 820, FDA Quality System Regulation. If the medical device under consideration is subject to only FDA regulatory requirements, and is exempt from Design Controls, the entire clause 7.3 in ISO 13485:2016 can be excluded from the QMS. However, if this medical device is also subject to other regulatory jurisdictions across the world, this may not be permissible.
Once again in Clause 1: Scope;
“If any requirement in Clauses 6, 7 or 8 of this International Standard is not applicable due to the activities undertaken by the organization or the nature of the medical device for which the quality management system is applied, the organization does not need to include such a requirement in its quality management system.”
The concept of not applicable, therefore, applies selectively to specific requirements of Clauses 6, 7 and 8. Clause 6 is Resource Management, Clause 7 is Product Realization and Clause 8 is Measurement, analysis and improvement. It is highly unlikely that a all of resource management, product realization or measurement, analysis and improvement can be excluded from any QMS. Therefore, only select, specific requirements from these clauses can be non-applicable in a given situation.
This the main reason for separating exclusion from non-applicability. Whereas, the entire Design and Development (Clause 7.3) can be excluded from the QMS, if permitted by applicable regulatory requirements, only select requirements of Clauses 6,7,8 can be excluded from the QMS based on non-applicability.
As an example, clause 7.5.5 – Particular requirements for sterile medical devices does not apply to an external service provider (e.g. contract manufacturer) who does not sterilize a medical device before sending it back to the original manufacturer. Similarly, third party software developer may not need to include certain requirements related to work environment (Clause 6.4.1) and contamination control (Clause 6.4.2) in the QMS.
The key point in understanding non-applicability is to clearly define the role of the organization, and the purpose of the QMS. Clause 4.1.1 requires that “the organization shall document the role(s) undertaken by the organization under the applicable regulatory requirements”. These roles can vary across the supply chain, from supplier to manufacturer to importer or distributor. As part of defining the role of the organization, an assessment of exclusion and non-applicability of ISO 13485:2016 requirements are expected to be documented in the quality manual (Clause 4.2.2)
Now let us look at the topic of Planning for Product Realization
The terms “plan”, “planned” or “planning” appear nearly 40 times in the normative part of ISO 13485:2016 compared to about 30 times in the 2003 version. As a result of a stronger emphasis on risk, there is also a stronger emphasis on planning and plans. In fact, risk is implicit whenever the requirements specify planning or a plan.
Clause 7 specifies requirements for Product Realization, and the very first sub-clause, is 7.1 - Planning of Product Realization. Specifically, “the organization shall plan and develop the processes needed for product realization.” Additionally, there is a requirement that “the organization shall document one or more processes for risk management in product realization.” The link between risk management in product realization and planning is quite clear in these requirements.
The confusion arises when the requirement of planning is also explicitly specified in sub-clause 7.3.2 – Design and Development Planning. Why have planning in two different sub-clauses within the same clause? Is there a difference between planning for product realization, and planning for design and development?
It is important to note that the term planning is used in the context of processes needed for product realization. It is expected that there will be planning across all processes as shown in the figure below:
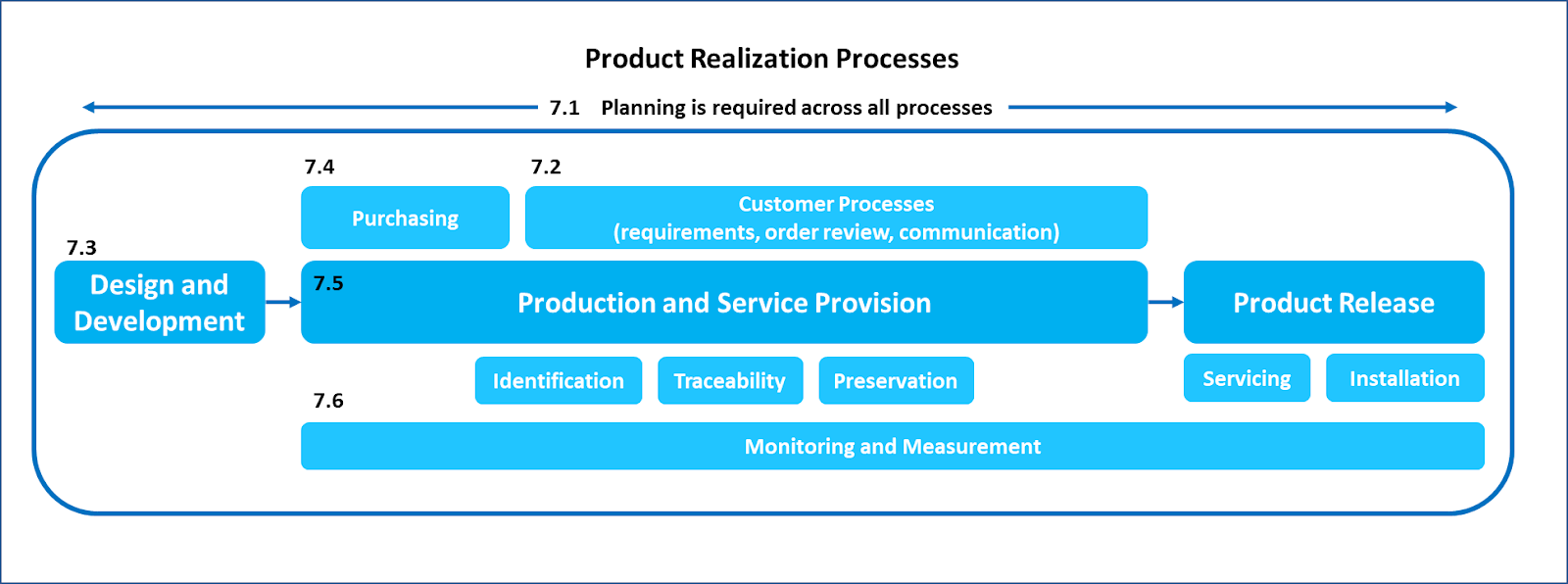
Design and Development is a sub-process within the overall Product Realization process. It is possible that design and development is done by a third party on behalf of the manufacturer; however, it is still considered to be an essential part of product realization. Design planning is critical for success in consistently meeting customer requirements. Therefore, the planning requirement is explicitly specified in sub-clause 7.3.2.
It is also possible that the entire sub-clause 7.3 – Design and Development is excluded from the QMS, if permitted by applicable regulatory requirements. In that case, planning would still be an expectation across other processes required for product realization. That is why there is a high-level requirement for planning in sub-clause 7.1 explicitly specified in the standard.
Bottom line – planning is an important requirement of the standard, which is expected to be visible in the QMS and supported by objective evidence.
How do you show objective evidence for planning? Planning needs to address the basic what, who, how, when and with what questions relevant for any activity or process. It can be demonstrated by procedure and/or practice. The standard does not specify how to plan, it simply specifies a requirement. Implicit in the requirement is a risk-based approach, which can be applied to determine the level of detail and control required in the plan for each individual process.
A significant challenge in understanding the requirements of ISO 13485:2016 is the fact that many of the terms are not defined within the standard. You need to refer to ISO 9000:2015 – Quality Management Systems – Fundamentals and Vocabulary, to fully understand the nature and intent of each requirement.
In conclusion, it is important to clearly understand the subtle, yet important, difference between the definition of exclusion and not-applicable when defining the scope of your Quality Management System. It is also important to apply a risk-based approach to planning the key processes involved in your product realization process and be able to provide objective evidence of such planning.
Share your comments and questions below. We are also available to provide customized training tailored to your specific situation. Contact us and let us know how we can help.