
A Pathway to Faster FDA Approvals
FDA is expanding the Abbreviated 510(k) Program with a Safety and Performance-based Pathway for certain medical devices.

All medical devices marketed in the United States are regulated by the FDA. These devices go through a rigorous regulatory review before they are cleared or approved for market release. There are two main pathways for this review – the pre-market notification, 510(k), and the pre-market approval (PMA). The 510(k) process results in device clearance, while the PMA process results in device approval. The 510(k) process is based on the concept of substantial equivalence. This means that the device under review should be equivalent in safety and effectiveness to another legally marketed predicated device, typically in a similar application and intended use.
Often, device manufacturers have to generate a lot of comparative data through direct testing against a predicate device to show substantial equivalence. In many situations, where the device type and intended use are well-understood, this type of testing may not be necessary. FDA recognizes that substantial equivalence in these cases can be demonstrated through a least burdensome regulatory approach, which is defined as the minimum amount of information necessary to adequately address a relevant regulatory question or issue through the most efficient manner at the right time.
In short, the idea is to drive efficiency through the regulatory process so new, innovative medical devices can be quickly introduced in the marketplace.
One-way FDA is encouraging a more efficient and faster device clearance is through the Abbreviated 501(k) Program. However, as shown in the Figure below, only a small fraction of the thousands of 510(k) submissions cleared each year go through the Abbreviated Program, while most go through the Traditional 510(k) Program. There is a third, Special 510(k) Program, applicable for well-defined device modifications, where a manufacturer modifies its own legally marketed device. We will focus only the Abbreviated and the Traditional 510(k) Programs in this article.

Consistent with the concept of least burdensome, devices cleared through the Abbreviated Program seem to take less time than the Traditional Program. As shown in the Figure below, 510(k) clearances through the Abbreviated Program in 2018 took around 20 fewer calendar days for submissions from both US and OUS (outside of the US) manufacturers. Prior years also show a similar difference in median days to decision between these two programs. Note that it is important to segment the data based on the country code because there is a significant difference in the days to decision. Also, note that the total time for the decision is dependent on processing times at the FDA as well as response times from the manufacturer. It is likely that the OUS manufacturers take longer to follow up on FDA requests for information and resolve outstanding issues.

Manufacturers may use the Abbreviated Program for their 510(k) submissions if they can show substantial equivalence through FDA guidance documents, special controls, and voluntary consensus standards. It is possible that not all devices may be eligible; however, the potential efficiency gain and shorter time frame for clearance is a great opportunity for the industry.
FDA is now expanding the Abbreviated Program through a recently announced Safety and Performance Based Pathway, applicable for a limited number of device types where substantial equivalence can be shown using FDA defined performance criteria. These performance criteria represent a level of performance similar to one or more existing, legally marketed devices based on the device type. The idea is that if a legally marketed device performs at certain levels relevant to its safety and effectiveness, and a new device meets or exceeds those performance levels, then FDA could find that the new device is as safe and effective without a direct, side-by-side, comparison.
As an example, metallic bone screws and washers used in orthopedic surgeries (product codes HWC, HTN) may be cleared through the safety and performance pathway if mechanical bench testing data on torsional yield strength, driving torque and axial pull out strength meet or exceed the FDA specified performance levels. Such performance criteria are defined in specific draft guidance documents for each eligible device type available on the FDA website (see references below).
Currently, there are 5 different product codes eligible for this pathway, but this list is expected to grow over time. A summary of the currently eligible devices is shown in the table below.
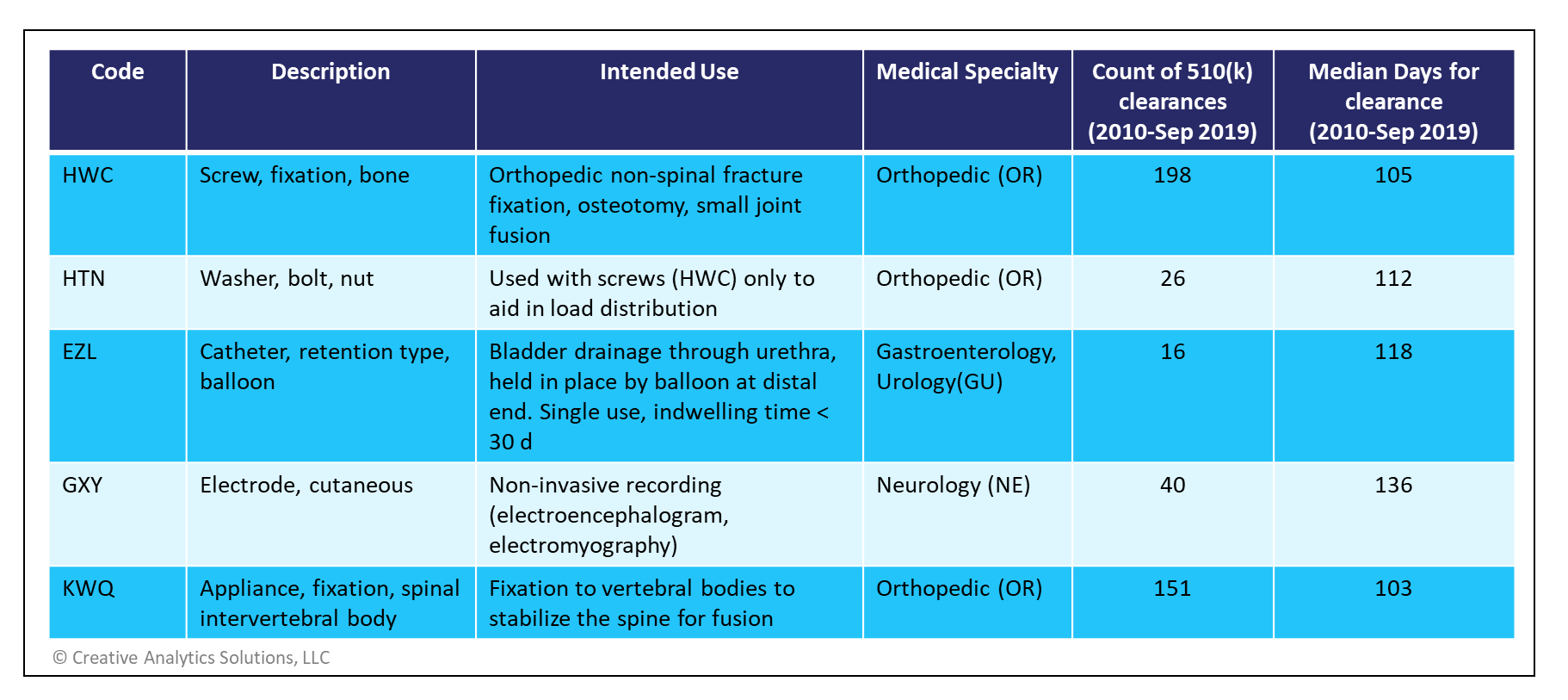
There is relatively little historical data on these device types. In recent years, only two submissions were cleared through the Abbreviated Program for devices under the GXY product code. One from Canada was cleared in 67 days while another from the US took 42 days. Nevertheless, these examples show a significant reduction in decision times compared to those submitted under the Traditional Program.
In conclusion, the Abbreviated 510(k) Program offers an excellent opportunity to drive significant efficiency gains in the regulatory submission and review process. Draft guidance from the FDA on certain product codes eligible for the Safety and Performance Based Pathway offer clear performance criteria, which should help streamline testing and data gathering for 510(k) submissions. Additional product codes are expected to be added to the list of eligible products in the future. FDA is also open to providing feedback on questions related to device eligibility for this pathway.
This is an exciting opportunity for the medical device industry!
References:
The Abbreviated 510(k) Program, September 2019
Safety and Performance Based Pathway, September 2019
Device-Specific Guidance for Safety and Performance Based Pathway, September 2019